
Regulatory
Biostatistics and Reporting are a critical part to any regulated clinical trial. We have mastered the use of RStudio within a regulatory, 21 CFR Part 11 compliant, framework to support Phase I and Phase II clinical trials as a Statistical Programmer and Regulatory Biostatistician.
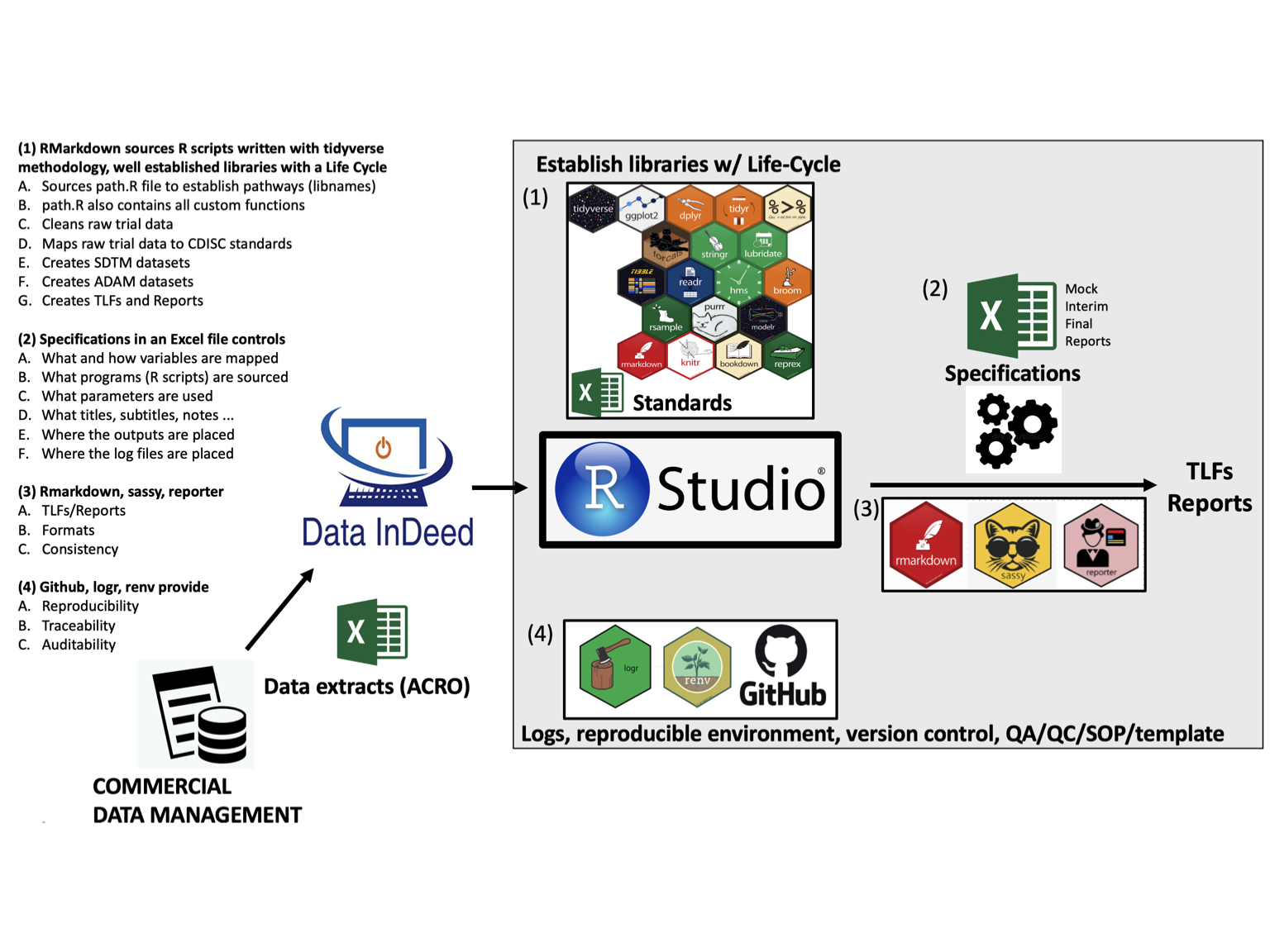
We support these major areas:
- Study Design and Protocol Development
- Statistical Consulting
- Sample Size Calculations
- Randomization Schema
- Statistical Analysis Plans (SAP)
- Data Safety and Monitoring Board (DSMB) Support
- Routine Clinical Trial/Sponsor Reports (i.e., weekly visits, events, dispositions and other safety reporting)
- Subject-level Listings
- Tables, Listings and Figures (TLFs)
- CDISC Compliant SDTM, ADAM, and Define.XML Files
- Clinical Study Reports
- Programming, Logs, and SOPs
Study Design and Protocol Development
We are experienced with the knowledge to develop optimal trial designs and write protocols for clinical development programs. From phase I/II Pharmokentic (PK) and Pharmacodynamic (PD) studies, early-phase dose-finding safety and efficacy studies to phase II trials, we can address your biostatistics needs and recommend the best solutions for your study.
We can provide oversight of clinical development programs as members of clinical / scientific advisory boards, clinical trial Steering Committees and DSMBs. We’ve supported submissions to FDA as part of routine duties while in the Army, participated in meetings with Investigational New Drug (IND) and Investigation Device Exemption (IDE) protocol discussions, and presented results of previous studies.
Statistical Consulting
In a consulting role, we provide support to Study Planning, Protocol Development, and Clinical Advisory Groups: drug development, scientific, statistical & trial design consulting. Further, we provide literature reviews, re-analysis / meta-analysis of reference studies, review and analysis of preclinical studies (toxicity, PK), analysis in support of the safety and efficacy sections of Investigator Brochures (IBs). In addition to simulation studies, we provide study design, sample size/power calculation, statistical considerations, randomization, including adaptive methods. Finally, we support client representation or support at IND / pre-study regulatory meetings.
Sample Size Calculations
During the planning stages of a clinical study, we’ll work with your team to establish reasonable estimates for effect and hypotheses of interest that reflect the study objectives under the study design, per SOP. Based on the hypotheses, we will:
- Determine the number of subjects needed to detect a clinically meaningful difference at a required level of power
- Discuss the trade-off in power should budget considerations be a factor determining the number of subjects
- If prior knowledge regarding the endpoints of interest is limited or inaccurate, we can help design your study to allow for interim analyses and sample size recalculation to ensure that your study is not under/over powered.
Randomization Schemas
We can assist Sponsors with developing randomization schema that minimize the variability of the parameter of interest and avoid confounding from other factors, per SOP. We are experienced in the following types of randomization:
- Simple: randomize subjects to one of two treatment groups
- Block: randomize subjects into treatment groups in blocks to ensure balance in sample size across groups over time. The block size is a multiple of the number of treatment groups.
- Stratified: randomize subjects to achieve balance among the treatment groups in terms of covariates, e.g. subject’s baseline characteristics.
- Dynamic: also known as adaptive randomization. An example is the minimization method in which subjects are randomized to a particular treatment group based on specific covariates and previous assignments of subjects.
Statistical Analysis Plans
We have the knowledge and experience to write SAPs and develop corresponding Tables, Listings and Figure (TLF) shells, based on the protocol and electronic case report forms (eCRF), per SOP. SAPs can be written for:
- Interim analysis
- Final/main analysis
- DSMB meetings
- Manuscripts
- Integrated Summaries of Safety and Efficacy (ISS/ISE) analysis
- Post-hoc/exploratory analysis
- Meta-analysis
DSMB Support
We have the skills, knowledge and experience to support DSMBs in the following areas:
- Preparation of DSMB charters / manuals
- Data exports and analyses for DSMB meetings: blinded and unblinded data
- Participation in DSMB meetings
- Preparation of DSMB statistical reports (open and closed sessions)
- Presentation of DSMB results
- Documentation of open/closed sessions
- Unblinding requests
ISS/ISE
For ISS/ISE, we will:
- Develop a data specification plan to integrate (pool) safety and efficacy data across multiple studies within the program
- Implement the data specification plan to generate standardized datasets for analysis
- Develop the SAP and TLF shells to assess safety and efficacy at the program level
- Program the integrated safety and efficacy TLFs
With pooled ISS data, we will:
- Identify common related adverse events and serious adverse events
- Identify safety concerns that show a pattern across all studies
- Assess safety in subgroups of subjects, if applicable
With pooled ISE data, we will:
- Assess efficacy in subgroups of subjects (e.g. pediatric population)
- Assess efficacy of secondary endpoint across all studies which would not have been possible under single studies
- Explore inconsistency in results between studies
- Assess sensitivity of results
Clinical Data Interchange Standards Consortium (CDISC) Standards
CDISC is the standard for submitting clinical data to Regulatory Agencies. As a member of the CDISC organization, we maintain up-to-date knowledge on Study Data Tabulation Model (SDTM) implementation guidelines. Our SDTM services include developing SDTM datasets for ongoing studies or converting legacy databases to SDTM standards.
We utilize industry standards/references when creating SDTM datasets:
- CDISC SDTM Implementation Guide
- CDISC SDTM guidance
- SDTM Controlled Terminology
- Indication-specific SDTM specifications, if applicable
Our processes in SDTM development are as follows:
- Map raw data variables to SDTM domains (identify domains, required, expected, permissible and relationship variables)
- Create SDTM specification documents
- Program SDTM domains
- Validate SDTM domains
- Annotate eCRF
- Create Define.xml
- Create Study Reviewer’s Guide (SRG)
- Produce Submission Package
- Quality Control at each step of the process, per SOP
Standards for creating analysis ready datasets are based on guidelines published by CDISC. We develop the specifications for Analysis Data Model (ADAM) datasets based on the SAP and the TLF shells. The source data for the ADAM datasets are preferably the SDTM datasets but it can also be done from the raw (native) study datasets.
We utilize industry standards/references when creating ADAM datasets:
- CDISC ADAM Implementation Guide
- CDISC ADAM OCCDS Guide
- ADAM Controlled Terminology
- Indication-specific ADAM domain specifications, if applicable
Our processes in ADAM development are as follows:
- Map relevant raw or SDTM data variables to ADAM domains
- Determine derived variables for analysis
- Determine analysis flags (population, sub-group, criterion-based) needed for analysis
- Create ADAM specification documents based on the above
- Program ADAM domains
- Validate ADAM domains
- Create Define.xml
- Create Study Reviewer’s Guide (SRG)
- Produce Submission Package
- Quality Control at each step of the process, per SOP
Finally, we have the experience and core competency in programming TLFs in support of:
- Final analyses
- Interim analyses
- DSMB meetings
- Annual safety updates / Development Safety Update Reports (DSURs)
- Integrated summaries of safety (ISS)
- Integrated summaries of efficacy (ISE)
- Abstracts/Manuscripts
- Post-hoc and exploratory analyses
The source data for TLF programming are the raw (native) study database, SDTM or ADAM datasets. All TLF programming is based on the shells created from the SAP. If shells are not available, we will create the shells upon request to ensure transparency and consistency of the output.
All programming is done by our statistical programmers per the SAP and TLF shell. Our SOPs define a separate program to be developed for each individual TLF. Programs can be shared directly with the agency upon request. We maintain logs for each study to define the extent and scope of quality control for TLFs. QC includes independent programming, and/or code review and/or content review of a subset of programs and/or TLF output. All issues are documented and resolved in the log.